Last update and review: August 28, 2021.
A short summary.
IL-10 is an anti-inflammatory cytokine. During infection it inhibits the activity of Th1 cells, NK cells, and macrophages, all of which are required for optimal pathogen clearance but also contribute to tissue damage. In consequence, IL-10 can both impede pathogen clearance and prevent immunopathology. Characteristic immunopathology of COVID-19 is a major factor implicated in severe forms of the disease and in “long COVID”.
Many different types of cells can produce IL-10, with the major source of IL-10 varying in different tissues or during acute or chronic stages of the same infection.
In a noteworthy 2008 article, – where there are some interesting insights but also quite serious errors, – Couper and coworkers, (1), reviewed the biology of IL-10, its cellular sources, and its role in viral, bacterial, and protozoal infections. Here, we share some of our notes taking during a literature review that included the article by Couper and coworkers (1).
Couper et al., 2008 (1) on IL-10 as the “Master Regulator” of Immunity of immune response to infection..
Inconsistencies in the nomenclature for regulatory T cell and the paucity of markers to differentiate them.
Couper et al., 2008:
“Sources of IL-10 during infection.
It is evident that IL-10 can be produced by many different myeloid and lymphoid cells and that more than one population of IL-10-producing cells may be induced during a single infection (Table I and Fig. 1). The complexity of IL-10 responses, together with inconsistencies in the nomenclature for regulatory T cell (Treg) populations (natural Treg (nTreg), Tr1, Th3, adaptive Treg, IL-10-producing T cells) and the paucity of markers to differentiate them, has resulted in considerable confusion in the published literature regarding the role of particular cell populations during specific infections.”
Irrespective of the source of IL-10, its effects are similar in all of the infections that have been studied: IL-10 suppresses macrophage and DC function, thereby limiting Th1 and Th2 effector responses.
Couper et al., 2008:
“Irrespective of the source of IL-10, its effects are similar in all of the infections that have been studied: IL-10 suppresses macrophage and DC function, thereby limiting Th1 and Th2 effector responses (Table I). Nevertheless, the impact of IL-10 is clearly determined by the timing and site of its production, and these are both likely to be affected by which cells are making IL-10. Moreover, because IL-10 production by one cell population can affect the ability of other cells to make IL-10, there is the potential for IL-10-producing cells to regulate each other. The picture that is emerging is that in infections of low to moderate virulence, i.e., inducing commensurately low to moderate inflammatory responses, IL-10 from DC or macrophages drives the production of IL-10 by nTreg, preventing pathology but allowing long-term escape of pathogens from immune control and giving rise to persistent (typically asymptomatic) infections; examples include C. albicans and mycobacterial infections. Alternatively, in highly virulent infections giving rise to strong proinflammatory responses, IL-10 production from larger populations of induced (adaptive) Treg seems to be required to minimize pathology during the resolution phase of the infection; the ability of Th1 cells to coproduce IL-10 and IFN-gamma may favor simultaneous pathogen clearance and suppression of downstream pathologies; examples include T. gondii, malaria, and leishmaniasis (17, 37, 64, 68). We postulate that the strength of the regulatory IL-10 response reflects the strength of the preceding inflammatory response (i.e., IL-6, IL-12, IL-23, and/or IL-27 levels) and that IL-10 from nTreg alone is insufficient to counter the very high levels of inflammation associated with virulent infections. Irrespective of the source of IL-10 (nTreg or Tr1), its production is of potential benefit to both the host (limiting pathology) and the pathogen (allowing persistent infection and thereby favoring onward transmission). However, if the source and thus the timing of IL-10 secretion are inappropriate, i.e., produced too early during a virulent infection or too late during an avirulent infection, overwhelming infection or severe tissue damage, respectively, will result (Fig. 2).”Some passages from Couper et al., 2008 (1), cited further, are somewhat misleading. Indeed, a shift from Th1 and Th17-meidated immune responses towards Th2 cells is definitely a part of the resolution of inflammation phase of the immune response. Notably, a shift from Th1 to Th2-type cells is observed when, in a paracrine fasion, immune cells start t converting the less active metabolite of vitamin D into the most active form of vitamin D (“calcitriol,” “1,25-dihydroxy vitamin D” or “1,25(OH)2D”). For example, Bruce et al., 2010 (2):
“Vitamin D and 1,25(OH)2D inhibit T helper type 1 (Th1)/Th17-mediated immune responses and autoimmune diseases by acting on the innate and acquired immune system to inhibit the function of Th1 and Th17 cells. Th1 and Th17 cells are important in host resistance to many infections including tuberculosis (TB) caused by Mycobacterium tuberculosis.”
Some passages from Couper et al., 2008 (1), cited further are somewhat misleading.
“The Th1:Th2 paradigm of immunity to infection in which Th1 cells were regarded as proinflammatory and Th2 cells were regarded as anti-inflammatory has undergone a major realignment in the last 10 years and, as in the development of the original paradigm, studies of infection have been crucial to this process. Depending on the nature of the infectious agent, both Th1 and Th2 cells are now known to mediate inflammation and tissue damage as well as pathogen killing. Anti-inflammatory functions, in contrast, are now known to rest with populations of regulatory cells that may be myeloid or lymphoid in origin and can include fully mature, classical Th1 or Th2 cells that produce IL-10 as a negative feedback mechanism to limit their own response. More than any other cytokine, IL-10 is an essential component of this regulatory response in almost all infections. Therapeutic strategies augmenting IL-10 to reduce host injury during infection must therefore be combined with effective antimicrobials to ensure that the underlying problem, the pathogen, is eliminated. Similarly, adjuvant therapies designed to block IL-10 or components of its signaling pathway in order to augment the effects of antimicrobial drugs or vaccines need to be applied with care, because they may have unintended side effects.”
“Table I”: Immunoregulatory roles of IL-10 during infection.
The following table from Couper et al., 2008 (1) is of interest, but it is somewhat misleading. Indeed, it is not the cytokine IL-10 alone that is responsible for the described effects on infections. Rather, the entire immune response evolves in a certain way, with immune cells engaging in an “inflammation resolution program” in a more or less appropriate and coordinated manner. Or, failing to engage due to complex interactions with pathogens as and other causes which results in a chronic inflammatory condition.

“Figure 1”: The sources and targets of IL-10 during infection.
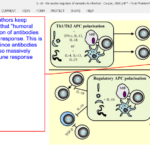
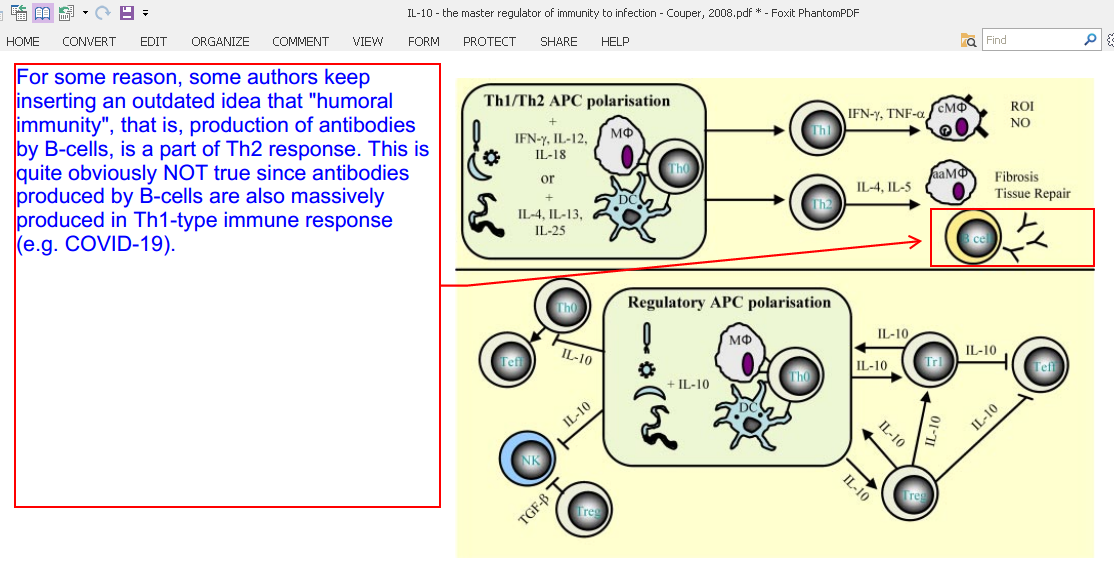
For some reason, some authors keep inserting in their papers an outdated idea that “humoral immunity”, that is, production of antibodies by B-cells, is a part of Th2 response. This is quite obviously NOT true since antibodies produced by B-cells are also massively produced in Th1-type immune response (e.g. COVID-19).
Couper et al., 2008:
“Interactions between pathogens and APC direct the development of Th1, Th2, or Treg populations depending on the cytokine environment and the level of costimulation. APC promoting regulatory responses are induced by priming under regulatory conditions (often involving IL-10 signaling) or as a result of direct modulation of their functions by the pathogen. IL-10 may act in an autocrine manner to suppress APC proinflammatory responses, may act directly on effector T cells to limit their proliferation and function, or may promote the differentiation of naive T cells into regulatory populations. Adaptive and natural regulatory T cells may then directly influence macrophage (MOˆ), DC, and effector T cell differentiation and effector functions via IL-10-dependent mechanisms. aaMO ˆ, Alternatively activated macrophage; cMOˆ , classical macrophage; ROI, reactive oxygen intermediate.”
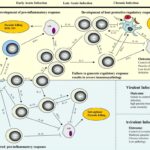
“Figure 2”: The source, timing, and magnitude of IL-10 production determines the outcome of protozoan infections.
Couper et al., 2008 (1):
“Early priming and polarization of proin-flammatory immune responses are crucial for the effective control of protozoal infection. Proinflammatory cytokine production by APC after interaction with pathogens and the subsequent activation of NK cells and Th1 cells are required to activate macrophages (MO) to eliminate microbes. Regulatory DC and macro-phages and/or regulatory T cells (Tr1, IL-10-Th1, and nTreg) must then control inflammation to prevent immune-mediated pathology. By contrast, early polar-ization toward regulatory, IL-10-producingAPC populations inhibits inflammation. In the case of avirulent (non-virulent) infections, this will allow the pathogen to persist in the absence of tissue damage. In the case of virulent infections, this leads to uncontrolled pathogen dissemination and death. ROI, Reactive oxygen intermediate.”
Conclusions.
Having a good grasp, staying current, deepening of our understanding of how immune system works is indispensable for maintaining good health and success in treating diseases.
Selected references:
1. Couper et al. J Immunol 2008; 180:5771-5777.
2. Bruce et al. Exp Biol Med (Maywood). 2010 August ; 235(8): 921–927.